
Trámite orientado a otorgar la Autorización de Ensayos Clínicos que se realizan con la intención de descubrir o verificar los efectos clínicos, farmacológicos y/o cualquier otro efecto farmacodinámico del producto en investigación,con el fin de asegurar el bienestar de los individuos participantes del Ensayo Clínico. Dicha autorización es otorgada a persona natural o jurídica.
Ensayos Clínicos: es cualquier investigación que se realice en seres humanos con intención de descubrir o verificar los efectos clínicos, farmacológicos y/o cualquier otro efecto farmacodinámico de producto(s) en investigación y/o identificar cualquier reacción adversa a producto(s) de investigación y/o para estudiar la absorción, distribución, metabolismo y excreción de producto(s) en investigación, con el objeto de comprobar su seguridad y/o eficacia.
Producto en investigación: Es toda forma farmacéutica de un ingrediente activo o placebo que se está probando o usando como referencia en un estudio o ensayo clínico.
El trámite Solicitud para la Aprobación de Ensayos Clínicos tiene como beneficiario a toda razón social que cuente con Registro Único de Contribuyentes (RUC) de persona natural o jurídica, nacional o extranjera, públicas o privadas.
Dirigido a: Persona Jurídica - Privada, Persona Jurídica - Pública, Persona Natural - Ecuatoriana, Persona Natural - Extranjera.
Aprobación de Ensayos Clínicos
1.-Solicitud dirigida a la máxima autoridad de la Agencia Nacional de Regulación, Control y Vigilancia Sanitaria- ARCSA por parte del patrocinador (ver formatos)
2.-Contrato entre el patrocinador y la Organización de Investigación por Contrato – OIC, debidamente suscrita por ambas partes;
3.- Ficha descriptiva del ensayo clínico (ver formatos)
4.-La aprobación del protocolo de ensayo clínico por parte de un Comité de Ética de Investigación en Seres Humanos (CEISH) que esté oficialmente reconocido por el Ministerio de Salud Pública.
a) Debe figurar además la declaración de no tener conflicto de interés por parte del comité en dicho ensayo clínico.
b) En el caso que el ensayo clínico se realice con sujetos en investigación a personas con discapacidad, se debe demostrar: que la investigación responda a las necesidades y prioridades de salud de la población con discapacidad y no puede realizarse en otros grupos; que la investigación esté diseñada para minimizar los riesgos de la afección que padece y que los beneficios sean superiores a los riesgos inherentes al estudio; que en los casos de discapacidad mental, en el proceso de obtención del consentimiento informado, las personas hayan recibido información del estudio clínico adaptada a su capacidad de comprensión, garantizando su participación en la medida de sus posibilidades y que el consentimiento informado sea otorgado por su representante legal.
c) En el caso que los ensayos clínicos que se realicen en adultos mayores, personas con discapacidad, personas privadas de libertad y quienes adolezcan de enfermedades catastróficas y de alta complejidad, por ser considerados grupos de atención prioritaria o vulnerables, deben ser de interés específico y limitarse a
aquellos que por su naturaleza solo puedan realizarse en este grupo poblacional.
d) En el caso de mujeres en edad reproductiva que hayan consentido participar en un estudio clínico, previo al inicio del mismo se realizará la confirmación de que la participante no esté embarazada.
5.-El protocolo de investigación, previamente autorizado por el Comité de Ética de Investigación en seres Humanos – CEISH, el cual debe estar firmado y sellado por el CEISH.
a) El protocolo de investigación, en el caso que el estudio de investigación se realicen en pueblos y nacionalidades del Ecuador, debe incluir la descripción y justificación del procedimiento a utilizar para comunicar la información a los sujetos en investigación.
b)En el caso que el ensayo clínico requiera muestras biológicas, en el protocolo debe especificar que dichas muestras son exclusivamente para el ensayo para el que el
sujeto otorga su consentimiento.
6.- El manual del investigador, según lo establecido en las Buenas Prácticas Clínicas
7.-El formato de consentimiento informado correspondiente al cual se agregará la versión del protocolo de investigación presentado, que haya sido previamente aprobado por un Comité de Ética de Investigación en Seres Humanos, el cual debe estar firmado y sellado por el CEISH. Teniendo en cuenta lo siguiente:
a) El documento de consentimiento informado debe contener la información detallada en el (ver formatos) del “Reglamento para la aprobación, desarrollo, vigilancia y
control de los Ensayos Clínicos”.
b) El documento de consentimiento informado y cualquier otro material informativo, que en relación al estudio sea proporcionado a los sujetos en investigación, deberá ser revisado y aprobado previamente por el Comité de ética de Investigación que aprobó el protocolo.
c) El documento de consentimiento informado no podrá contener ninguna expresión que dé lugar a que el sujeto que consiente en el mismo, exima de responsabilidad
por negligencia, o por riesgos derivados del ensayo clínico.
d) En el caso de personas analfabetas, se registrará su huella digital en el documento de consentimiento informado y firmarán como testigos dos personas que el participante del estudio elija, siempre y cuando no tengan conflicto de interés con la investigación que se desarrollará y no estén vinculadas de ninguna manera con
el equipo de investigación.
e) En el caso de estudios clínicos realizados en pueblos y nacionalidades del Ecuador, a más del consentimiento informado individual, se deberá realizar una consulta comunitaria previa, y se presentará una copia del documento de aprobación por parte de las autoridades de la comunidad.
8.-El informe farmacológico de aprobación de comercialización del producto en investigación emitido por la ARCSA, o número de Registro Sanitario con el nombre comercial del producto; si fuera el caso de estudios post-comercialización.
9.- Las cartas de declaración de interés institucional y disponibilidad del establecimiento de salud, para la realización del ensayo clínico, emitida por la máxima autoridad de los
establecimientos que sean propuestos como centros de investigación clínica.
10.-La carta compromiso suscrita por el investigador principal de cada centro de investigación clínica participante en el ensayo clínico, en la que se señale lo siguiente:
a) Que participará en calidad de investigador en el estudio,
b)Que ha sido capacitado sobre el protocolo de la investigación y que por tanto lo conoce y está a conformidad con el mismo,
c) El valor monetario que recibirá por sus servicios,
d)Así como la determinación de sus responsabilidades en el estudio.
11.- La lista del centro o centros de investigación clínica, que incluya el detalle de todos los investigadores y su equipo de trabajo por centro, que participarían en el ensayo en el país, especificando el tipo de establecimiento de salud, títulos profesionales de cada integrante y su rol en el estudio.
12.- Inscripción de los investigadores principales del estudio, en la base de datos del Ministerio de Salud Pública a cargo de la Dirección Nacional de Inteligencia de la Salud, o quien ejerza sus competencias, en tanto la Secretaría Nacional de Educación Superior, Ciencia, Tecnología e Investigación - SENESCYT desarrolle esta herramienta y coordine con el Ministerio de Salud Pública la existencia de una base de datos única de investigadores.
13.- La hoja de vida del investigador principal y de sus colaboradores por cada centro de investigación clínica que evidencie experiencia en la especialidad que se estudia; y capacitación en desarrollo de ensayos clínicos, Buenas Prácticas Clínicas y temas afines.
14.-El cronograma del estudio a desarrollarse en el país: el cronograma debe especificar como mínimo los siguientes aspectos:
a) Fecha de inicio del ensayo clínico a nivel mundial (cuando aplique)
b) Fecha tentativa de inicio del ensayo clínico en el país
c) Número de sujetos previstos para reclutar en el estudio a nivel mundial
d) Número de sujetos previstos para reclutar en el estudio en el país
e) Fecha tentativa para someter a aprobación del EC por el CEISH
f) Fecha tentativa para someter a aprobación por parte de la ARCSA
g) Fecha del taller de inicio del estudio
h) Fecha de convocatoria y toma de consentimientos informados
i) Fecha tentativa de reclutamiento del primer paciente
j) Calendario de visitas programadas para los pacientes
k) Calendario de toma de muestras para exámenes complementarios
l) Fecha tentativa de solicitud para importación del medicamento en investigación
m) Fecha tentativa de solicitud para kits de laboratorio
n) Fecha de envíos de informes de avance del estudio (anual)
o) Fecha de envío de informes de seguridad del estudio (anual)
p) Fecha de cierre del estudio a nivel mundial (cuando aplique)
q) Fecha tentativa de visita del último paciente en el país
r) Fecha tentativa de presentación del informe final del estudio al CEISH correspondiente y la ARCSA.
s) Fecha tentativa de publicación de resultados
15.-Un certificado del sistema de gestión de calidad del laboratorio clínico o de la institución en la que se encuentra el laboratorio clínico que participará en el estudio.
16.- El detalle del producto en investigación y otros medicamentos a utilizarse en el ensayo (ver formatos); así como de la etiqueta del producto en investigación y del embalaje.
Las etiquetas del producto en investigación deberán especificar en su envase primario, con tinta indeleble en idioma castellano, la siguiente información: Nombre del promotor, Título del ensayo clínico, Nombre del producto en investigación, cuando aplique, Fecha de vencimiento, Código del producto, Código del protocolo, Número de unidades, Número de lote, Forma farmacéutica, Vía de administración, Condiciones de almacenamiento, conservación y utilización, La leyenda: “Producto para uso exclusivo en investigación, prohibida su venta”. En el caso de envases primarios que por su espacio no permitan establecer esta información, se imprimirá la misma en un envase secundario.
17.-Informes de evaluación de riesgo sanitario en las fases previas de los estudios (Fase I, II y III), para que la ARCSA establezca el balance riesgo-beneficio del producto en investigación, se presentarán los estudios preclínicos del producto de investigación.
18.- La certificación de Buenas Prácticas de Manufactura de la planta de fabricación del medicamento en investigación, dicha certificación debe ser emitida por la Autoridad Competente del país donde se fabrica el producto, y en el caso que el producto sea fabricado o acondicionado en varios países se debe adjuntar el Certificado de BPM de cada una de las plantas que intervienen en la fabricación del producto; dicho certificado debe estar apostillado o consularizado según corresponda.
19.- La certificación de lotes del medicamento en investigación, con ficha estabilidad en condiciones de humedad y temperatura correspondiente a la zona climática IV o de acuerdo a las condiciones que requiera el producto; y caducidad del producto en investigación.
20.- El presupuesto general del ensayo (ver formatos).
21. Copia del contrato celebrado entre el investigador principal y el promotor.
23. En los casos de que los centros de investigación clínica se encuentren ubicados en establecimientos de salud de la Red Pública Integral de Salud (RPIS), se deberá presentar copia del convenio entre el establecimiento de salud y el promotor, mismo que como mínimo debe contener los siguientes consideraciones:
a) En los establecimientos de salud de la RPIS en los que se realicen ensayos clínicos, los costos directos e indirectos generados por la realización del ensayo, serán cubiertos en su totalidad por el patrocinador del mismo;
b)Todas las atenciones asistenciales que se realicen en un establecimiento de salud público, en las fases de reclutamiento, ejecución y posibles complicaciones que se presten durante o después de terminado el ensayo, serán cubiertas por el patrocinador del ensayo conforme al Tarifario de Prestaciones del Sistema Nacional de Salud vigente;
c) Los estímulos que reciban los investigadores que trabajan en establecimientos de la RPIS por sus servicios prestados, en la ejecución de un ensayo clínico, que se desarrollen en un establecimiento de este tipo, fuera de su jornada laboral, deberán constar en el convenio que suscriba el promotor y el establecimiento de salud de la RPIS;
d) En el convenio, adicionalmente constará la autorización del establecimiento de salud para que el investigador pueda utilizar las instalaciones del establecimiento
para efectos del ensayo clínico, fuera de su jornada laboral.
24.- La lista de suministros necesarios para el desarrollo del ensayo clínico.
25. El cuaderno de recogida de datos.
26. El flujograma de manejo de eventos adversos y reacciones adversas.
27. El formulario de notificación de sospecha de reacciones adversas graves inesperadas y/o evento adverso grave (ver formatos)
28. El compromiso juramentado del patrocinador de entregar el informe final del ensayo a la ARCSA.
29. Copia de la póliza de seguro de responsabilidad civil emitida por una compañía de seguros establecida en el Ecuador, facultada para el efecto. La póliza cubrirá las responsabilidades de todos los implicados en la investigación, así como las del centro de investigación clínica en el que se realice el ensayo clínico.
La cobertura deberá abarcar la ejecución del ensayo clínico y se extenderá al menos un (1) año después de finalizado éste, a fin de cubrir las consecuencias que se demuestren sean derivadas del ensayo clínico en cuestión. Luego de aprobado un ensayo, si éste dura más de un (1) año, el promotor presentará a la ARCSA un compromiso de renovación de la póliza, al menos con tres (3) meses de anticipación a su caducidad; y presentará la póliza renovada una vez que sea emitida.En el caso que el patrocinador es un individuo que investigue bajo contrato con la academia, será ésta la que contrate la póliza de seguro para los riesgos derivados de la investigación que realice.
30. En el caso de los productos de referencia que se utilicen en el estudio clínico que sean importados de otro país, se debe adjuntar la copia apostillada del Registro Sanitario o Certificado de Comercialización, emitido por la Autoridad Competente del país donde se comercializa el producto.
Notas:
Los Centros de Investigación a participar en el Ensayo Clínico deben estar inscritos en el Registro Nacional de Centros de Investigación Clínica de la Agencia Nacional de Regulación, Control y Vigilancia - ARCSA.(Art. 87 Reglamento para la aprobación, desarrollo, vigilancia y control de los Ensayos Clínicos- Acuerdo Ministerial Nro 0075)
El investigador Principal deberá ser un profesional de la salud con título de cuarto nivel, relacionado con el área a investigar. (art. 90 Reglamento para la aprobación, desarrollo, vigilancia y control de los Ensayos Clínicos- Acuerdo Ministerial Nro 0075)
1.- En los casos que aplique, se entregará la delegación del patrocinador para el desarrollo del ensayo clínico en el país a una sola Organización de Investigación por Contrato, mediante un contrato o convenio legalizado en el que consten las obligaciones de cada una de las partes.
2.- Traducción certificada y apostillado o legalizada de documentos en otro idioma diferente al español.
Presencial
1.-El patrocinador o la Organización de Investigación por Contrato – OIC, debe ingresar la solicitud dirigida a la máxima autoridad de la ARCSA con copia a la Dirección Técnica de Ensayos Clínicos o quien ejerza sus competencias, adjuntando todos los requisitos en físico y digital (archivos en formato PDF, grabados en un CD), a la ARCSA por Secretaria General de Planta Central o a través de las Coordinaciones Zonales, (ver FORMATOS).
2.-Revisar el sistema de Quipux, mediante el cual se realiza la entrega de la orden de pago de acuerdo a los valores establecidos en Resolución ARCSA-DE-2018-019-JCGO, la cual deberá ser cancelada conforme a lo declarado en el instructivo externo GE-C.4.3.5-TAS-01 Guía para el pago de importes por los servicios brindados por la ARCSA.
3.- Revisar el sistema de Quipux dentro de cinco (5) días calendario posteriores a la recepción de la documentación, para verificar si los documentos entregados estan completos y correctos.
4.- El solicitante (patrocinador u Organización de Investigación por Contrato - OIC, según corresponda), debe subsanar las observaciones emitidas por la ARCSA en treinta (30) días término, mediante Quipux. De no recibir respuesta del usuario en el tiempo establecido se cancelará el trámite automáticamente. El solicitante tiene máximo dos oportunidades para subsanar las observaciones.
5.-Una vez que la ARCSA verifique que toda la información y requisitos estén completos y correctos se considerará como admitido el trámite, con lo cual se emitira por QUIPUX la entrega de la orden de pago de acuerdo a los valores establecidos en Resolución ARCSA-DE-2018-019-JCGO, la cual deberá ser cancelada conforme a lo declarado en el instructivo externo GE-C.4.3.5-TAS-01 Guía para el pago de importes por los servicios brindados por la ARCSA.
6.- La ARCSA realizará una revisión técnica detallada de la documentación presentada diez (10) días término, si existen observaciones la ARCSA notifica al solicitante mediante Quipux para la respectiva subsanación.
7.- El solicitante tiene quince (15) días témino para subsanar las observaciones emitidas por la ARCSA, en el caso de no subsanar dichas observaciones en el término establecido se cancelará el trámite.
8.- Una vez subsanadas todas las observaciones, la ARCSA en cinco (5) días témino, elabora el informe técnico respectivo.
9.- La ARCSA remite el informe técnico mediante Quipux al Comité Técnico Asesor para Investigación Clínica, y convoca en un plazo de diez (10) días días témino al Comité para evaluación del Ensayo Clínico.
10.- La ARCSA analiza el resultado de la evaluación del Comité Técnico Asesor para Investigación Clínica, y elabora en cinco (5) días témino, el informe de evaluación del protocolo de investigación y determina la aprobación, no aprobación o ampliación de la información respecto al ensayo clínico.
11.- La ARCSA comunica al solicitante la aprobación, no aprobación o ampliación de la información respecto al Ensayo Clínico, mediante Quipux.
Canales de atención: Presencial, Sistema de Gestión Documental Quipux (www.gestiondocumental.gob.ec).
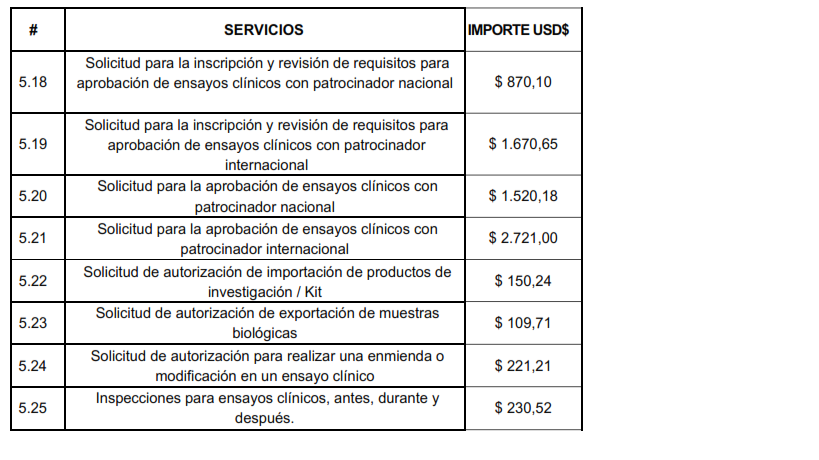
Solicitud para la inscripción y revisión de requisitos para aprobación de ensayos clínicos con patrocinador nacional $ 870,10
Solicitud para la inscripción y revisión de requisitos para aprobación de ensayos clínicos con patrocinador internacional $ 1.670,65
Solicitud para la aprobación de ensayos clínicos con patrocinador nacional $ 1.520,18
Solicitud para la aprobación de ensayos clínicos con patrocinador internacional $ 2.721,00
ARCSA Planta Central (Ciudadela Samanes, Av. Francisco de Orellana y Av. Paseo del Parque, Parque Samanes, Bloque 5, Guayaquil - Ecuador). Código Postal: 090703.
De lunes a viernes 08h00 a 17h00.
9 Coordinaciones Zonales https://www.controlsanitario.gob.ec/contacto/
Contacto: Dirección Técnica de Atención al Usuario
Email: atencionalusuario@controlsanitario.gob.ec
Teléfono: 043727440
Art. 6.- Para la realización de un ensayo clínico en el Ecuador se requiere que toda persona natural o jurídica, pública, privada, nacional o extranjera que sea patrocinador de la realización del ensayo, solicite la aprobación previa a la Agencia Nacional de Regulación, Control y Vigilancia Sanitaria-ARCSA.
Art. 8.- Para autorizar un ensayo clínico, la ARCSA solicitará al patrocinador del estudio la presentación, al menos de la siguiente documentación:
a) Una ficha descriptiva del ensayo clínico (Anexo 1.)
b) La aprobación del protocolo de ensayo clínico por parte
de un Comité de Ética de Investigación en Seres
Humanos (CEISH), oficialmente reconocido por el
Ministerio de Salud Pública.
c) En los casos que aplique, se entregará la delegación del
patrocinador para el desarrollo del ensayo clínico en el
país a una sola Organización de Investigación por
Contrato, mediante un contrato o convenio legalizado en
el que consten las obligaciones de cada una de las
partes.
d) El protocolo de investigación.
e) El manual del investigador, según lo establecido en las
Buenas Prácticas Clínicas.
f) El formato de consentimiento informado correspondiente
al cual se agregará la versión del protocolo de
investigación presentado, que haya sido previamente
aprobado por un Comité de Ética de Investigación en
Seres Humanos.
g) El informe farmacológico de aprobación de
comercialización del producto en investigación emitido
por la ARCSA, si fuera el caso de estudios postcomercialización.
h) Las cartas de declaración de interés institucional y
disponibilidad del establecimiento de salud, para la
realización del ensayo clínico, emitida por la máxima
autoridad de los establecimientos que sean propuestos
como centros de investigación clínica.
i) La carta compromiso suscrita por el investigador principal
de cada centro de investigación clínica participante en el
ensayo clínico, en la que se señale que participará en
calidad de investigador en el estudio, que ha sido
capacitado sobre el protocolo de la investigación y que
por tanto lo conoce y está a conformidad con el mismo,
el valor monetario que recibirá por sus servicios, así
como la determinación de sus responsabilidades en el
estudio.
j) La lista del centro o centros de investigación clínica, que
incluya el detalle de todos los investigadores y su
equipo de trabajo por centro, que participarían en el
ensayo en el país, especificando el tipo de
establecimiento de salud, títulos profesionales de cada
integrante y su rol en el estudio.
k) Inscripción de los investigadores principales del estudio,
en la base de datos del Ministerio de Salud Pública a
cargo de la Dirección Nacional de Inteligencia de la
Salud, o quien ejerza sus competencias, en tanto la
Secretaría Nacional de Educación Superior, Ciencia,
Tecnología e Investigación - SENES CYT desarrolle
esta herramienta y coordine con el Ministerio de Salud
Pública la existencia de una base de datos única de
investigadores.
l) La hoja de vida del investigador principal y de sus
colaboradores por cada centro de investigación clínica
que evidencie experiencia en la especialidad que se
estudia; y, capacitación en desarrollo de ensayos
clínicos, Buenas Prácticas Clínicas y temas afines.
m) El cronograma del estudio a desarrollarse en el país.
n) Un certificado del sistema de gestión de calidad del
laboratorio clínico o de la institución en la que se
encuentra el laboratorio clínico que participará en el
estudio.
o) El detalle del producto en investigación y otros
medicamentos a utilizarse en el ensayo, conforme el
modelo que consta en el Anexo 2 de este Reglamento,
así como de la etiqueta del producto en investigación y
del embalaje.
p) Informes de evaluación de riesgo sanitario en las fases
previas de los estudios (Fase I, II y III), para que la
ARCSA establezca el balance riesgo-beneficio del
producto en investigación.
q) La certificación de Buenas Prácticas de Manufactura de
la planta de fabricación del medicamento en
investigación.
r) La certificación de lotes del medicamento en
investigación, con ficha estabilidad y caducidad.
s) El presupuesto general del ensayo, según el modelo que
consta en el Anexo 3 de este Reglamento.
t) Copia del contrato celebrado entre el investigador
principal y el promotor.
u) En los casos de que los centros de investigación clínica
se encuentren ubicados en establecimientos de salud
de la Red Pública Integral de Salud, se deberá
presentar copia del convenio entre el establecimiento de
salud y el promotor.
v) La lista de suministros necesarios para el desarrollo del
ensayo clínico.
w) El cuaderno de recogida de datos.
x) El flujograma de manejo de eventos adversos y
reacciones adversas.
y) El formulario de notificación de sospecha de reacciones
adversas graves inesperadas y/o evento adverso grave
del ensayo clínico.
z) El compromiso juramentado del patrocinador de entregar
el informe final del ensayo a la ARCSA.
aa) Copia de la póliza de seguro de responsabilidad civil
emitida por una compañía de seguros establecida en el
Ecuador, facultada para el efecto. La póliza cubrirá las
responsabilidades de todos los implicados en la
investigación, así como las del centro de investigación
clínica en el que se realice el ensayo clínico. La
cobertura deberá abarcar la ejecución del ensayo clínico
y se extenderá al menos un (1) año después de
finalizado éste, a fin de cubrir las consecuencias que se
demuestren sean derivadas del ensayo clínico en
cuestión. Luego de aprobado un ensayo, si éste dura
más de un (1) año, el promotor presentará a la ARCSA
un compromiso de renovación de la póliza, al menos
con tres (3) meses de anticipación a su caducidad; y
presentará la póliza renovada una vez que sea emitida.
La póliza de responsabilidad civil deberá sujetarse a las
disposiciones del Código Orgánico Monetario y Financiero,
Libro III Ley General de Seguros, Código de Comercio y
demás normativa vigente sobre la materia; y contendrá, al
menos, lo siguiente: el título del estudio, duración del
estudio, cobertura de la atención requerida, tipos de riesgos
asegurados, pruebas clínicas cubiertas, valor límite de
cobertura al asegurado por evento y el monto de la póliza, el
cual será determinado según el riesgo de la investigación
clínica que conste en el protocolo aprobado.
Artículo 5.- Los importes a cobrarse por los servicios, derivados para la aprobación o autorización de Ensayos Clínicos, será los siguientes:
| # | Año-Mes | Volumen de Quejas | Volumen de Atenciones |
|---|---|---|---|
| 1 | 2025-01 | 0 | 0 |
| 2 | 2025-02 | 0 | 1 |
| 3 | 2025-03 | 0 | 0 |
| 4 | 2025-04 | 0 | 0 |
| 5 | 2025-05 | 0 | 1 |
| 6 | 2025-06 | 0 | 2 |
| 7 | 2024-01 | 0 | 0 |
| 8 | 2024-02 | 0 | 0 |
| 9 | 2024-03 | 0 | 0 |
| 10 | 2024-04 | 0 | 0 |
| 11 | 2024-05 | 0 | 0 |
| 12 | 2024-06 | 0 | 0 |
| 13 | 2024-07 | 0 | 0 |
| 14 | 2024-08 | 0 | 0 |
| 15 | 2024-09 | 0 | 0 |
| 16 | 2024-10 | 0 | 0 |
| 17 | 2024-11 | 0 | 0 |
| 18 | 2024-12 | 0 | 1 |
| 19 | 2023-01 | 0 | 0 |
| 20 | 2023-02 | 0 | 0 |
| 21 | 2023-03 | 0 | 0 |
| 22 | 2023-04 | 0 | 0 |
| 23 | 2023-05 | 0 | 0 |
| 24 | 2023-06 | 0 | 0 |
| 25 | 2023-07 | 0 | 0 |
| 26 | 2023-08 | 0 | 0 |
| 27 | 2023-09 | 0 | 0 |
| 28 | 2023-10 | 0 | 0 |
| 29 | 2023-11 | 0 | 0 |
| 30 | 2023-12 | 0 | 0 |
| 31 | 2022-01 | 0 | 0 |
| 32 | 2022-02 | 0 | 0 |
| 33 | 2022-03 | 0 | 2 |
| 34 | 2022-04 | 0 | 1 |
| 35 | 2022-05 | 0 | 1 |
| 36 | 2022-06 | 0 | 3 |
| 37 | 2022-07 | 0 | 0 |
| 38 | 2022-08 | 0 | 0 |
| 39 | 2022-09 | 0 | 0 |
| 40 | 2022-10 | 0 | 0 |
| 41 | 2022-11 | 0 | 1 |
| 42 | 2022-12 | 0 | 1 |
| 43 | 2021-01 | 0 | 2 |
| 44 | 2021-02 | 0 | 1 |
| 45 | 2021-03 | 0 | 1 |
| 46 | 2021-04 | 0 | 0 |
| 47 | 2021-05 | 0 | 0 |
| 48 | 2021-06 | 0 | 0 |
| 49 | 2021-07 | 0 | 0 |
| 50 | 2021-08 | 0 | 0 |
| 51 | 2021-09 | 0 | 0 |
| 52 | 2021-10 | 0 | 0 |
| 53 | 2021-11 | 0 | 0 |
| 54 | 2021-12 | 0 | 0 |
| 55 | 2020-01 | 0 | 1 |
| 56 | 2020-02 | 0 | 0 |
| 57 | 2020-03 | 0 | 0 |
| 58 | 2020-04 | 0 | 0 |
| 59 | 2020-05 | 0 | 0 |
| 60 | 2020-06 | 0 | 0 |
| 61 | 2020-07 | 0 | 0 |
| 62 | 2020-08 | 0 | 0 |
| 63 | 2020-09 | 0 | 0 |
| 64 | 2020-10 | 0 | 0 |
| 65 | 2020-11 | 0 | 2 |
| 66 | 2020-12 | 0 | 1 |
| 67 | 2019-01 | 0 | 0 |
| 68 | 2019-02 | 0 | 0 |
| 69 | 2019-03 | 0 | 0 |
| 70 | 2019-04 | 0 | 0 |
| 71 | 2019-05 | 0 | 0 |
| 72 | 2019-06 | 0 | 0 |
| 73 | 2019-07 | 0 | 0 |
| 74 | 2019-08 | 0 | 0 |
| 75 | 2019-09 | 0 | 0 |
| 76 | 2019-10 | 0 | 0 |
| 77 | 2019-11 | 0 | 0 |
| 78 | 2019-12 | 0 | 0 |
| 79 | 2017-12 | 1 | 4 |
Fecha de última actualización: 2022/04/27